河南省-科技部共建细胞分化调控国家重点实验室培育基地
State Key Laboratory Cultivation Base for Cell Differentiation Regulation

过氧化物酶体是机体必需的多功能细胞器,涉及ROS代谢、脂质氧化、缩醛磷脂和胆汁酸的合成等,在氧化还原、脂质代谢、炎症通路调控中发挥关键作用。随着研究不断深入,过氧化物酶体缺陷与过氧化物酶体生物发生障碍、神经退行性疾病、病毒感染性疾病、癌症及其他疾病的发病进程相关。尽管相关研究揭示了过氧化物酶体对相关疾病的功能和作用机制,但其对于损伤后修复再生的具体机制仍需探索。
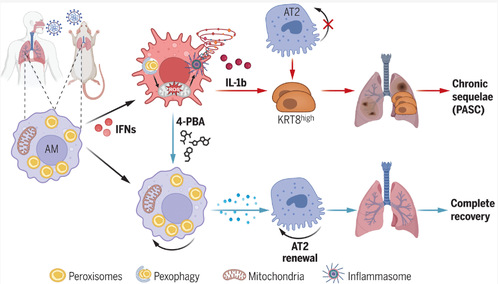
2025年3月7日,弗吉尼亚大学医学院孙杰团队在Science杂志上发表了题为Macrophage peroxisomes guide alveolar regeneration and limit SARS-CoV-2 tissue sequelae的研究论文。该研究揭示了COVID-19损坏巨噬细胞中的过氧化物酶体,导致肺修复受损、长期炎症和慢性纤维化。肺泡巨噬细胞中的过氧化物酶体在促进肺泡再生、抑制异常炎症及防止肺纤维化中具有关键作用,并提出了基于过氧化物酶体靶向治疗新冠急性后遗症的新策略。
通过生信分析发现,与不同物种的其他肺细胞类型相比,肺泡巨噬细胞(AM)表达较高水平的过氧化物酶体相关基因;与其他组织巨噬细胞相比,肺巨噬细胞在人和小鼠中均具有最高的过氧化物酶体基因表达水平。基于此,进一步对比健康供体和COVID-19患者的肺泡灌洗液单细胞转录组数据、肺组织蛋白表达情况和组织免疫荧光,发现在COVID-19感染的患者中肺巨噬细胞的过氧化物酶体数量显著下降,但形状变大且出现异常聚集。同时,在小鼠SARS-CoV-2和IAV呼吸道病毒感染模型中所得结果与人病例中观察到的现象相符。因此,病毒感染可能引起肺巨噬细胞中过氧化物酶体生物发生改变、区室重塑,导致其功能损伤。
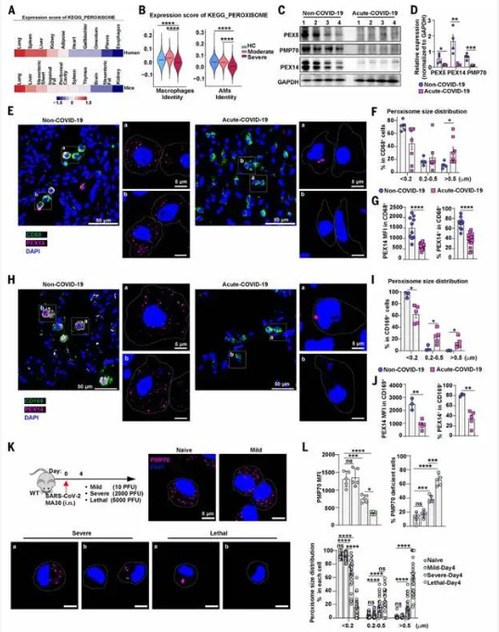
2.干扰素抑制过氧化物酶体生物发生并促进巨噬细胞中的过氧化物酶体自噬
为了研究病毒感染驱动肺巨噬细胞中过氧化物酶体重塑的机制,研究使用可以触发呼吸道病毒感染的几种信号物质刺激小鼠AM,通过WB和免疫荧光实验发现IFNα或IFNγ处理导致过氧化物酶体数量减少且聚集体增加,其中IFNγ诱导后变化更明显。同时,SARS-CoV-2感染小鼠后使用中和抗IFNγ和抗IFNα/β受体(IFNAR)分别阻断IFNα和IFNγ信号传导,阻断IFNγ导致过氧化物酶体PMP70 水平增加,且AM中过氧化物酶体聚集体减少。此外,IFNγ处理增加了PMP70 泛素化,触发过氧化物酶体的选择性自噬。因此,IFNγ通过抑制过氧化物酶体生物发生和促进过氧化物酶体降解引发过氧化物酶体重塑。
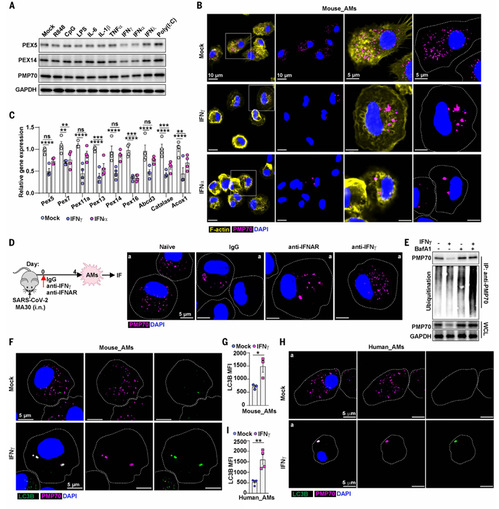
3.巨噬细胞过氧化物酶体是炎症消退所必需的
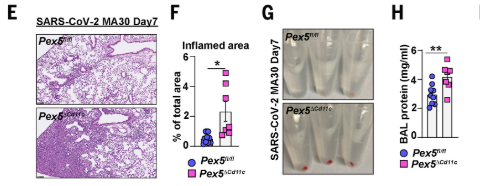
为了探讨在病毒感染后肺巨噬细胞中过氧化物酶体的病理生理功能,作者利用CD11ccrePex5Flox/Flox (Pex5ΔCD11c)和 Lyz2crePex5Flox/Flox (Pex5ΔLyz2)小鼠研究肺巨噬细胞中过氧化物酶体缺失前后的表型变化。感染呼吸道病毒后,统计小鼠生存率,基因缺失小鼠表现出更高的发病率及较低的生存率。同时,结合趋化因子测定、流式细胞分析以及肺组织病理学分析,结果表明过氧化物酶体功能缺失不会显著影响宿主的抗病毒免疫或急性炎症反应,但会阻碍病毒清除后的肺部炎症消退和组织损伤修复。

本研究通过整合单细胞RNA测序、免疫细胞亚群定量及功能实验,系统揭示了在SARS-CoV-2感染后过氧化物酶体缺陷通过肺泡巨噬细胞(AMs)调控肺部免疫-修复失衡的双重机制。首先,scRNA-seq与炎症通路富集分析显示,感染后Pex5缺陷小鼠肺部免疫微环境显著紊乱,且AT2和AMs数量锐减;基因层面表现为炎症信号过度激活,而细胞连接组装、上皮再生等修复通路受抑制。尽管AMs普遍上调免疫应答基因,但其修复功能(如生长因子分泌)因过氧化物酶体缺陷而丧失,导致免疫稳态失衡与组织再生障碍。
进一步通过体外伤口愈合、三维类器官培养及多组学分析发现,AMs过氧化物酶体对肺泡上皮修复具有关键调控作用。Pex5缺陷AMs在感染7天后上皮增殖和分化相关通路显著下调,伴随AT1和AT2细胞数量减少及Ki67+增殖标志降低。功能实验证实,正常AMs可通过分泌因子促进MLE-12迁移及AT2类器官形成,而缺陷AMs完全丧失此能力。因此,AMs过氧化物酶体通过调控巨噬细胞-上皮细胞互作,介导AT2细胞自我更新及肺泡上皮损伤后修复,其功能缺失导致病毒性肺损伤后再生障碍。
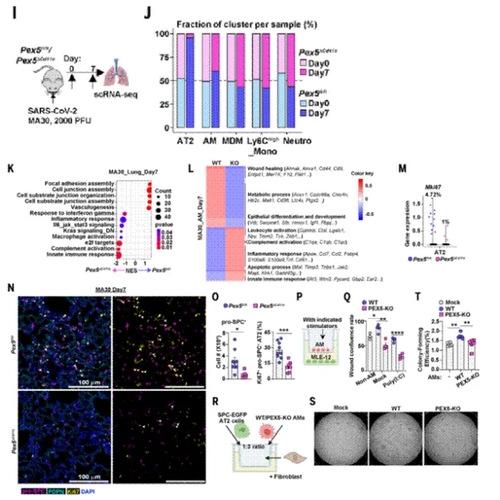
5.过氧化物酶体对脂质代谢及线粒体健康具有细胞类型特异性调控作用
为揭示过氧化物酶体在巨噬细胞的脂代谢及线粒体功能中具有细胞类型特异性调控机制。通过脂质组学分析发现过氧化物酶体缺陷导致肺泡巨噬细胞中极长链脂肪酸(VLCFAs)异常积累、醚脂合成减少,而骨髓来源巨噬细胞(BMDMs)受影响较小,提示过氧化物酶体功能具有巨噬细胞亚群特异性。此外,利用线粒体呼吸功能检测(OCR/ECAR)、线粒体膜电位及活性氧(ROS)测定等技术,进一步揭示了AMs中过氧化物酶体缺陷损伤线粒体功能,而BMDMs及中性粒细胞等髓系细胞的线粒体功能未受显著影响,表明过氧化物酶体作用具有组织特异性。同时,线粒体ROS积累是AMs修复基因(如上皮再生相关基因)表达下调的关键因素,使用线粒体ROS抑制剂后可恢复其促进伤口愈合能力。因此,过氧化物酶体缺乏可能通过破坏脂质代谢和线粒体健康来阻碍肺巨噬细胞的伤口愈合和促进修复功能。
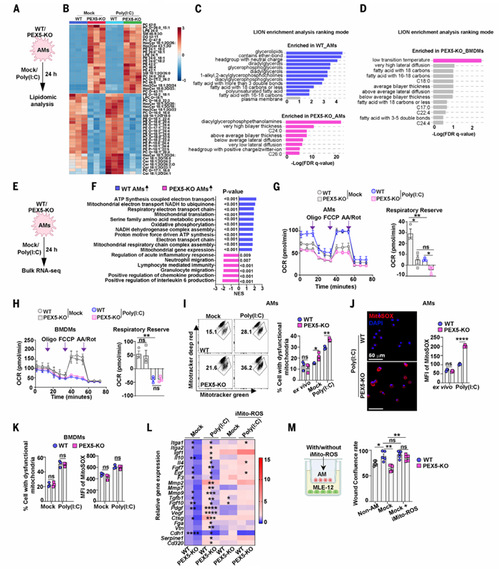
6.巨噬细胞过氧化物酶体可防止发育不良的过渡祖细胞积累及肺部后遗症形成
通过整合单细胞测序、拟时序分析与分子生物学实验,系统解析了肺泡巨噬细胞的过氧化物酶体缺陷导致肺泡上皮修复障碍的分子机制。首先,scRNA-seq与免疫组化显示,Pex5ΔCD11c小鼠感染SARS-CoV-2后,肺泡上皮分化轨迹异常,KRT8过渡祖细胞在感染后7天至21天持续积累,而成熟AT1和AT2细胞数量锐减,伴随肺纤维化及慢性炎症。进一步通过体外原代AT2细胞模型发现,过氧化物酶体缺陷的巨噬细胞因线粒体功能障碍激活NLRP3炎症小体,导致Caspase-1活性升高及IL-1β异常分泌,其条件培养基可抑制AT1分化基因,上调过渡细胞标志物,而IL-1β中和抗体可逆转此效应。此外,WB显示PEX5缺陷AMs中GSDMD-NT膜定位增强,焦亡增加。线粒体ROS抑制剂与GSDMD抑制剂可抑制IL-1β分泌,但无法阻断GSDMD裂解。因此,AMs过氧化物酶体通过维持线粒体稳态抑制炎症小体过度激活,其缺陷触发ROS-GSDMD-IL1β轴异常,导致肺泡上皮转分化阻滞及过渡细胞病理性滞留,最终促进肺纤维化。
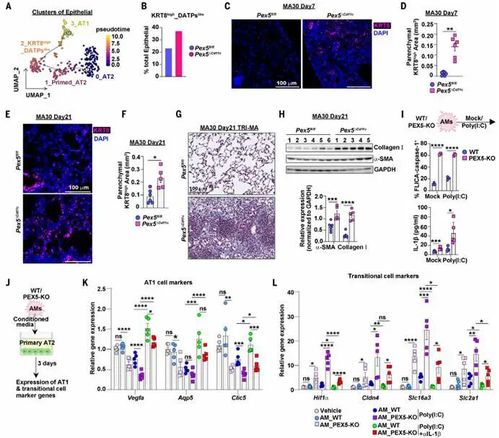
7.PASC肺组织中巨噬细胞过氧化物酶体持续性损伤
本研究通过免疫荧光染色、相关性分析及SARS-CoV-2感染小鼠模型,揭示了COVID-19后遗症相关肺纤维化(PASC-PF)中巨噬细胞过氧化物酶体持续性损伤与肺重塑异常的机制。临床样本分析显示,PASC-PF患者肺组织中含过氧化物酶体的巨噬细胞比例显著降低,而KRT8过渡祖细胞异常积累,且两者空间分布呈显著负相关,提示巨噬细胞代谢缺陷直接驱动异常上皮重塑。动物模型进一步验证:中老年雄性C57BL/6小鼠感染SARS-CoV-2后21天,肺组织呈现慢性炎症与纤维化,其MerTK+巨噬细胞过氧化物酶体水平持续减少,同时KRT8细胞比例升高,与临床表型高度一致。研究表明,病毒性肺损伤后,巨噬细胞过氧化物酶体功能障碍长期存在,其缺失通过驱动KRT8细胞滞留及纤维化进程,成为PASC-PF的核心病理机制。
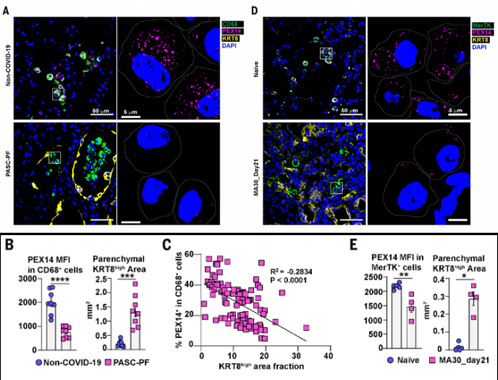
8.通过靶向过氧化物酶体生物合成来治疗PASC的潜在策略
研究通过药理学干预促进过氧化物酶体生物发生,系统评估其对病毒性肺损伤及慢性后遗症(PASC)的治疗潜力。4-苯基丁酸(4-PBA),作为已知可促进过氧化物酶体生成的小分子药物。4-PBA处理肺泡巨噬细胞,显著提升AMs过氧化物酶体蛋白表达,抑制过氧化物酶体自噬,逆转IFNγ诱导的代谢紊乱;在IAV或SARS-CoV-2感染小鼠急性期给予4-PBA,治疗后降低感染小鼠死亡率,减轻急性炎症反应及纤维化,并促进AT1/AT2细胞再生;在病毒清除后对中老年雄性小鼠实施延迟治疗,可减少KRT8细胞滞留、改善肺泡上皮数量,抑制慢性炎症与纤维化,且该效应依赖过氧化物酶体功能。4-PBA通过增强过氧化物酶体生物发生,恢复巨噬细胞代谢稳态,从而抑制病毒诱导的异常免疫应答与上皮重塑,有效缓解急性肺损伤并阻断慢性后遗症进展。
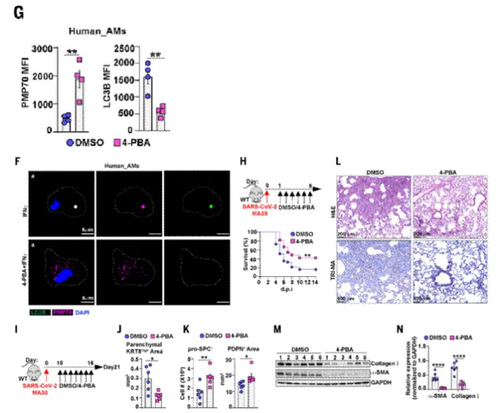
本研究揭示了巨噬细胞过氧化物酶体在调控病毒性肺损伤后炎症消退与组织再生中的作用。严重呼吸道病毒感染通过增强干扰素信号(尤其是IFN-γ),抑制过氧化物酶体生物发生并促进其自噬降解,导致巨噬细胞代谢功能障碍。过氧化物酶体缺失引发脂代谢紊乱与线粒体损伤,激活NLRP3炎症小体,驱动Gasdermin D孔形成及IL-1β异常释放,进而促进KRT8过渡上皮细胞病理性累积,阻碍AT2向AT1分化,最终引发慢性纤维化与肺功能不全。此外,通过药理学干预增强过氧化物酶体生成,4-PBA可恢复巨噬细胞代谢稳态,缓解急性炎症并阻断慢性后遗症进展。该研究提出靶向过氧化物酶体代谢通路是治疗病毒后肺损伤及PASC的潜在策略,为改善长期COVID患者的临床结局提供了新方向。
原文链接:https://www.science.org/doi/10.1126/science.adq2509?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed