河南省-科技部共建细胞分化调控国家重点实验室培育基地
State Key Laboratory Cultivation Base for Cell Differentiation Regulation

本研究整合单细胞RNA测序与空间转录组学,在IPF患者AT2细胞中定位FAO通路基因(如CPT1a)时空表达缺陷。借助3D类器官模型与iPSC源AT2细胞,发现CPT1a抑制剂etomoxir诱导AT2细胞分化为促纤维化过渡态细胞。构建AT2细胞特异性CPT1a敲除小鼠及双损伤模型,证实FAO缺陷加剧肺纤维化,通过代谢动态分析和谱系追踪揭示FAO缺失致线粒体功能障碍及SMAD7乙酰化水平下降,激活TGF-β信号通路,驱动SASP和胶原沉积。同时发现年龄依赖性FAO活性降低是IPF易感关键因素,补充乙酸盐可恢复SMAD7稳定性并逆转过渡态细胞分化,建立“代谢缺陷-表观修饰失调-纤维化激活”调控轴,为相关治疗策略及代谢干预提供新方向。

美国俄亥俄州立大学内科学系肺病、重症监护与睡眠医学分部的Ana L.Mora团队在JCI Insight上发表了题为“Regulation of lung progenitor plasticity and repair by fatty acid oxidation”的研究论文,该论文揭示了FAO缺陷促进肺纤维化的重要机制,为靶向FAO或乙酰CoA代谢治疗IPF提供了理论依据。
1.FAO基因在衰老和IPF肺上皮细胞中的表达下降
研究团队分析了衰老和IPF过程中肺上皮细胞中脂肪酸氧化酶(FAO)基因的表达情况。单细胞RNA测序显示,IPF患者的AT2细胞中脂质和线粒体代谢相关通路显著下调,且细胞群中FAO基因的表达量从健康老年人肺到IPF肺逐渐降低。空间转录组学证实IPF肺中SFTPC+细胞减少,且FAO基因(如CPT1a、ACADL)在SFTPC+细胞中表达降低。免疫荧光显示,IPF患者AT2细胞(HT2280+)中CPT1a蛋白表达显著低于年轻和健康老年供体。
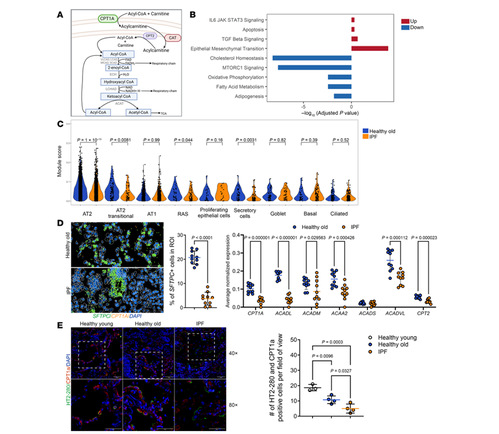
2.抑制CPT1a诱导人AT2类器官中基底样和分泌细胞表型
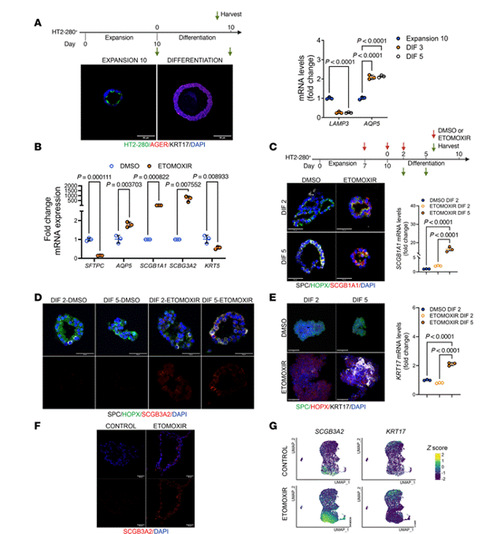
CPT1a抑制剂etomoxir处理的AT2类器官中,AT2标志物SFTPC减少,分泌标志物SCGB1A1、SCGB3A2和基底样标志物KRT17显著增加。分化阶段etomoxir处理进一步促进SCGB1A1、KRT17的表达,表明FAO抑制诱导AT2向异常中间态分化。iAT2(诱导多能干细胞来源的AT2)类器官中,etomoxir处理导致SCGB3A2和KRT17高表达,单细胞测序验证其异质性。
3.AT2细胞中CPT1a缺失增加肺纤维化易感性
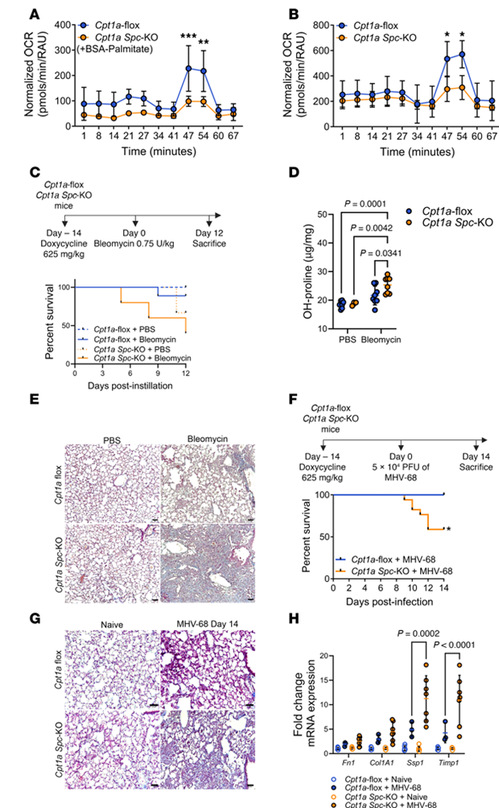
为了探究AT2细胞中FAO缺陷的生理后果,研究团队对CPT1a基因进行了敲除,发现CPT1a敲除小鼠的AT2细胞的FAO能力和线粒体呼吸显著降低。在博来霉素诱导的肺损伤中,CPT1a敲除小鼠死亡率升高,胶原沉积和纤维化标志物显著增加;MHV68病毒感染模型中,CPT1a敲除小鼠同样表现出更高的纤维化评分和促纤维化基因表达,表明在AT2细胞中CPT1a缺乏会导致损伤后更易发生肺纤维化。
4.CPT1a缺失促进肺泡分化中间态(ADI)细胞的出现
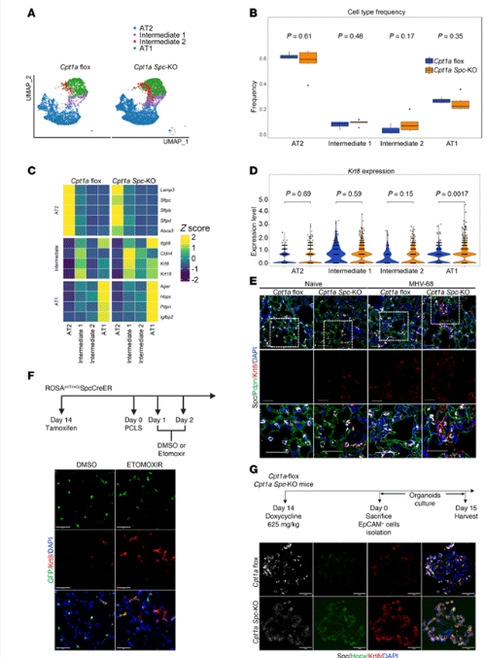
该团队继续研究了CPT1a基因缺失对小鼠肺上皮细胞的影响。单细胞RNA测序结果显示,CPT1a敲除小鼠的肺泡上皮细胞中出现了两种中间细胞群体,其中中间1亚群(Intermediate 1)具有ADI细胞的转录特征,如Krt8、Krt18等。此外,CPT1a缺失导致AT1细胞中Krt8表达显著增加,但AT1细胞的分化标志物Igfbp2也上调。通过免疫荧光和类器官培养实验进一步验证,发现CPT1a缺失促使未受伤和损伤后的肺部出现Krt8+中间态细胞,并且AT2细胞在分化过程中同时表达AT2、AT1和ADI细胞标志物,表明分化异常。
5.CPT1a缺失诱导呼吸肺泡分泌细胞(RAS)表型
在CPT1a缺陷的AT2细胞中,通过基因表达分析发现分泌细胞标记物Segb1a1基因显著上调。此外,中间2细胞群(Intermediate 2)在CPT1a敲除小鼠中表现出气道分泌细胞的特征,而AT2和ADI细胞标记物的表达较低。同时,这些细胞群在与上皮间质转化(EMT)、缺氧和胆固醇稳态相关的通路中富集,并且糖酵解相关基因表达增加。通过免疫荧光和谱系追踪技术验证了在CPT1a敲除小鼠的肺中,支气管周围和间质区域存在的Scgblal+细胞来源于AT2细胞,而在3D类器官培养中检测到更多的Scgbla1+细胞,进一步支持CPT1a缺陷促进AT2细胞向RAS表型转变的结论。
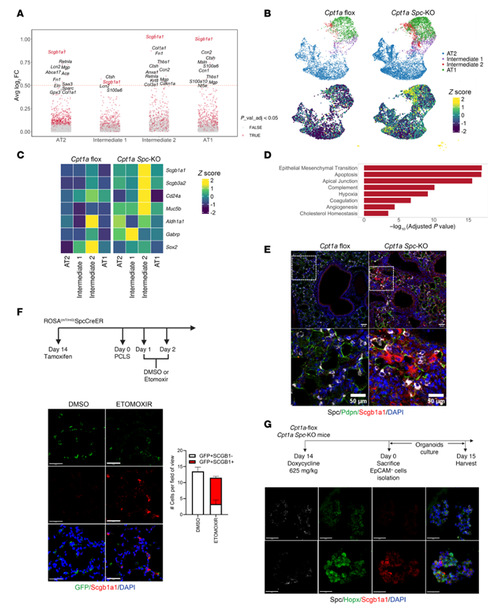
6.CPT1a缺失促进细胞衰老
CPT1a缺失会增加处于中间状态细胞的衰老标志物的表达。单细胞测序显示,CPT1a敲除小鼠所有上皮细胞群(AT2、ADI、AT1)中衰老标志物(如Cdkn1a、SASP基因)表达上调,衰老评分显著升高,表明CPT1a的缺失会导致衰老表型的建立。
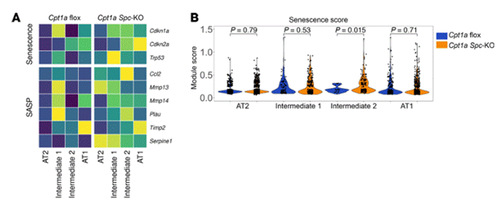
7.CPT1a缺陷导致线粒体功能障碍
此外,研究人员构建CPT1a基因敲除的小鼠肺上皮细胞模型MLE 12,揭示脂肪酸氧化(FAO)缺陷通过破坏代谢稳态和线粒体功能,诱导细胞衰老及病理表型。实验表明,CPT1a缺失导致长链脂肪酸氧化能力下降、脂滴堆积、线粒体复合物I活性降低及NAD+/NADH失衡,同时触发衰老标志物(如p21)和促炎因子(如IL-6)的表达上调,并激活缺氧、TGF-β等信号通路。这些变化与肺纤维化中异常中间态细胞的代谢紊乱和衰老特征一致,提示靶向恢复脂肪酸氧化或线粒体功能可能成为干预纤维化的新策略。
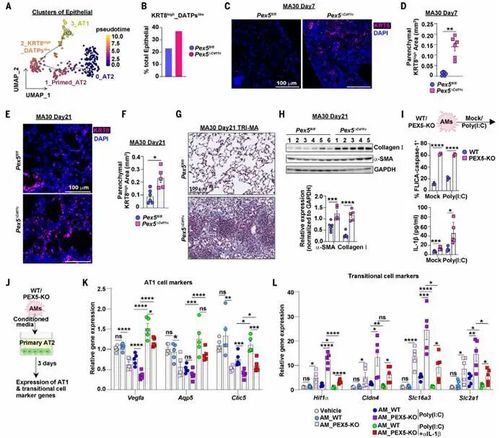
8.CPT1a缺陷激活TGFβ信号通路
在CPT1a缺陷小鼠(CPT1a Spc-KO)及人类AT2类器官模型中,肺泡上皮细胞(AT2、中间态2和AT1细胞)的TGF-β靶基因(如Tgfb、Serpine1、Fn1)表达显著上调,且单细胞测序显示TGF-β通路激活评分升高。机制上,CPT1a缺失导致脂肪酸氧化(FAO)能力下降,乙酰辅酶A(acetyl-CoA)生成减少,进而抑制TGF-β抑制因子SMAD7的乙酰化修饰。SMAD7乙酰化不足使其泛素化降解增加,导致其蛋白水平下降,解除对SMAD2/3磷酸化的抑制,从而激活TGF-β信号。通过补充乙酸盐恢复乙酰辅酶A水平,可显著恢复SMAD7稳定性,并抑制TGF-β通路活性,降低中间态细胞标志物(KRT8、SCGB1A1)的表达。此外,CPT1a抑制剂etomoxir处理的类器官中,TGF-β诱导的AT2细胞向分泌型中间态分化也被乙酸盐干预逆转。
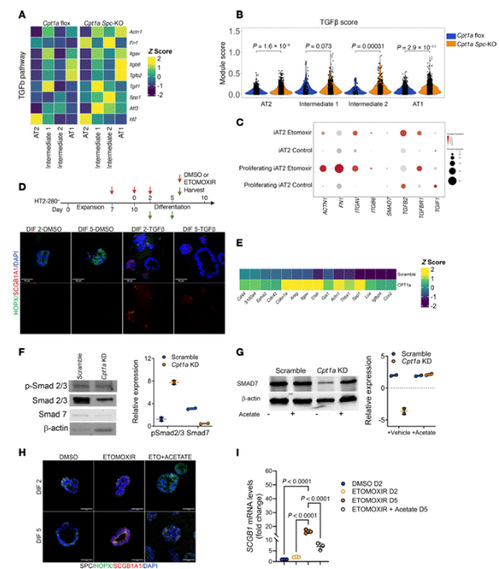
该研究揭示了脂肪酸氧化代谢通路在调节肺祖细胞修复反应中的重要作用,以及其在肺纤维化发展中的潜在机制。CPT1a缺乏最有可能通过扰乱乙酰辅酶A水平的调节来促进TGF-β信号通路的激活,从而导致AT2细胞分化为中间细胞状态。通过补充乙酸盐恢复乙酰辅酶A水平,抑制TGF-β通路,为肺纤维化提供了潜在的治疗靶点。
原文链接:
https://insight.jci.org/articles/view/165837